Vascular complications remain the leading cause of morbidity and mortality in individuals with diabetes, affecting critical organs such as the heart, kidneys (nephropathy), and vasculature (atherosclerosis), even in the context of intensive glucose control. While current therapies effectively manage metabolic dysregulation, they do not reverse established vascular injury. This residual risk has been attributed to metabolic memory, wherein transient episodes of poor glycaemic control can trigger long-lasting vascular damage that persists despite subsequent normoglycaemia.
Diabetic vascular dysfunction is driven by a triad of chronic hyperglycaemia, oxidative stress, and inflammation, with endothelial dysfunction serving as a central pathophysiological event. Emerging evidence implicates endothelial-to-mesenchymal transition (EndMT) as a key mechanism contributing to the loss of endothelial identity and acquisition of a pro-inflammatory, pro-fibrotic phenotype. This shift exacerbates vascular inflammation and fibrosis, accelerating the progression of atherosclerosis. Over the past decade, our team has identified the SET7 lysine methyltransferase as a critical regulator of hyperglycaemia-induced EndMT, endothelial inflammation, and oxidative stress — core drivers of diabetic vascular complications. Importantly, our recent studies demonstrate that genetic deletion of SET7 in diabetic mice markedly attenuates vascular injury. This is evidenced by a reduction in atherosclerotic plaque formation and improved renal endothelial integrity, including decreased albuminuria. These findings provide compelling evidence that endothelial cells are key sites of SET7 action, and that targeted inhibition of SET7 may offer a novel, epigenetically guided therapeutic strategy for mitigating diabetic vascular complications, as illustrated.
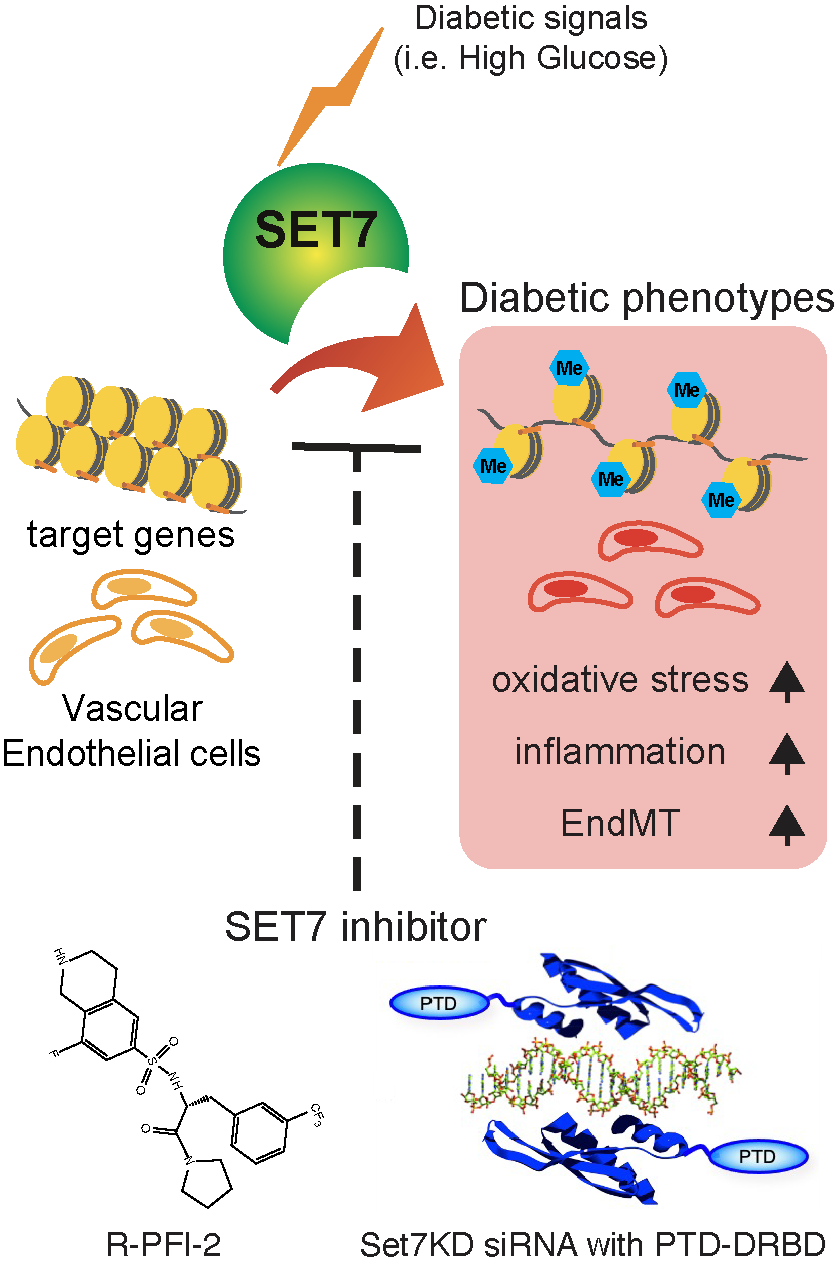 Inhibition of SET7 reverses hyperglycaemia-induced endothelial dysfunction in diabetes. High glucose activates SET7 in endothelial cells, leading to sustained expression of genes driving oxidative stress, inflammation, and EndMT. These changes promote vascular complications such as atherosclerosis and nephropathy. Targeted SET7 inhibition using R-PFI-2 or SET7KD siRNA with PTD-DRBD offers a novel approach to disrupt this epigenetic memory and reduce diabetic vascular injury.
Inhibition of SET7 reverses hyperglycaemia-induced endothelial dysfunction in diabetes. High glucose activates SET7 in endothelial cells, leading to sustained expression of genes driving oxidative stress, inflammation, and EndMT. These changes promote vascular complications such as atherosclerosis and nephropathy. Targeted SET7 inhibition using R-PFI-2 or SET7KD siRNA with PTD-DRBD offers a novel approach to disrupt this epigenetic memory and reduce diabetic vascular injury.
Metabolic memory — where vascular complications persist despite glycaemic control9-12 — underscores the need for therapies targeting underlying molecular drivers. Epigenetic programming is a key mechanism2-4,13. We have shown that hyperglycaemia activates SET7 in endothelial cells, inducing H3K4me and durable transcriptional changes5-7. SET7 also regulates histone and non-histone targets in stem cells14 and innate immunity15, linking it to sustained vascular injury in diabetes.
We aim to validate endothelial SET7 inhibition as a novel therapeutic strategy to prevent or reverse diabetic vascular complications.
- Assess the effects of SET7 inhibition in primary endothelial cells from individuals with type 1 diabetes, using both pharmacological and RNAi-based approaches to define epigenetic and inflammatory responses.
- Demonstrate the therapeutic efficacy of SET7 inhibitors in preclinical diabetic mouse models, evaluating vascular and renal outcomes relevant to human disease.
- Establish the role of endothelial-specific SET7 inhibition in attenuating vascular injury, to define cell-specific mechanisms and therapeutic relevance for complications such as atherosclerosis and diabetic nephropathy.